 |
Member
|
|
Join Date: Sep 2006
Posts: 782
|
|
|
Member
Join Date: Sep 2006
Posts: 782
|
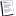
Statins and oxidative stress.
Oxidative stress is a result of altered balance in the relative concentrations of oxidants and antioxidants. Ox-LDL is deleterious to endothelial and VSMCs; it activates macrophages, induces release of various cytokines, and increases endothelial adhesiveness resulting in vascular injury and inflammation. Statins as potent antioxidants and antiatherosclerotic agents are attractive therapeutic options for preserving normal vascular function and blood flow. In several human and animal studies, various statins have been shown to: 1) inhibit the uptake and generation of ox-LDL (38), 2) attenuate vascular and endothelial superoxide anion formation by inhibition of NADH oxidases via Rho-dependent mechanisms (39, 40); and 3) preserve the relative levels of vitamin E, vitamin C, and endogenous antioxidants such as ubiquinone and glutatione in LDL particles (41, 42, 43). Thus, statins not only decrease oxidants but also restore antioxidants, thereby possibly reducing the level of oxidative stress in the vascular milieu, which may explain some of the observed clinical beneficial effects.
Statins and thrombosis.
Statins have been shown to play a role in altering the levels of several key elements in the process of thrombosis. Different statins have varying effects on prothrombotic factors, such as tissue factor, tissue factor pathway inhibitor, platelet aggregation, blood and plasma viscosity, fibrinogen, plasminogen activator inhibitor 1 (PAI-1), and lipoprotein (a) (44). Cellular expression of tissue factor in human macrophages is suppressed by lipophilic statins (45). Statins normalize thrombin generation in hypercholesterolemic patients and reduce platelet aggregation (46). Furthermore, decreases in platelet aggregation after statin therapy may be partially related to relative reductions in the cholesterol to phospholipid content in the platelet membrane (47).
Elevated fibrinogen levels and plasma viscosity may contribute to increased risk for CVD events in patients with and without established coronary artery disease (20, 46). Conflicting results exist on the effects of statins on fibrinogen levels and blood viscosity (48, 49, 50). Elevated plasminogen activator inhibitor 1 levels are associated with prothrombotic states, and statins reduce these levels (51); however, this effect is not a class effect. Further studies are needed to explain the apparent conflicting results.
Statins and vasculogenesis.
Statins, in addition to modulating endothelial and vascular function, may mediate neovascularization (vasculogenesis) and collectively contribute to the reduction in recurrent CVD events. Increased vasculogenesis has been demonstrated in rabbits treated with simvastatin via the activation of vascular Akt (21). Statins mobilize endothelial progenitor cells (EPCs) from the bone marrow that play a role in maintenance vasculogenesis (52). Increased EPCs are seen immediately after a coronary event (53) and line the endothelium of myocardial vessels (54). Indeed, statin therapy is associated with enhanced EPCs in patients with coronary artery disease (55).
Statins and kidneys
Statins have been shown to attenuate renal injury in both in vivo and in vitro studies. Renal injury initiates inflammatory cascades that involve similar cellular events as seen in vascular tissue. Statins inhibit key events in this process that alter the progression of renal injury. In hyperglycemic insulin-deficient diabetic rats, pravastatin ameliorates the structural and functional changes of diabetic nephropathy (56). Although the pathogenesis of diabetic nephropathy is complex and multifactorial, statins have been demonstrated to decrease TGF-ß production and suppress the enhanced Ras-dependent activation of MAPK cascade (Fig. 4Go). Lovastatin has similar action on glomerular disease in obese insulin-resistant rats (57). In another model of renal injury due to overexpression of Ang II, cerivastatin decreased systolic blood pressure, albuminuria, and cortical necrosis (28). These changes were associated with reduced infiltration of inflammatory cells, diminished expression of adhesion molecules, and lower levels of transcription factor (NF-{kappa}B) activity (Fig. 4Go). In rats with glomerulonephritis, simvastatin decreased mesangial cell proliferation and monocyte/macrophage infiltration (58). Statins have been shown to inhibit the proliferative actions of platelet-derived growth factor (59) and TGF-ß (56). Cytokines released during renal injury activate NF-{kappa}B and growth-regulating pathways in mesangial and tubular cells. Statins both decrease the levels of cytokines and inhibit the NF-{kappa}B-dependent gene activation, such as MCP-1 and IL-6. In humans, statins also decrease urinary albumin excretion in patients with nephrotic syndrome and in patients with type II diabetes (4). Thus, statins modulate glomerular mesangial and interstitial inflammatory process independent of lipid reduction. Clinical relevance of these observations is yet to be determined by the ongoing interventional studies.
Statins and glucose metabolism
A retrospective analysis of the WOSCOPS examining the development of new diabetes mellitus revealed that pravastatin therapy reduced the risk of developing diabetes by 30%. This prevention in the onset of diabetes was associated with significant reduction in triglyceride levels, but upon further analyses the reduction in triglycerides did not account for the effect of statins on the development of diabetes (5). Recent advances in understanding the cellular actions of statins may explain mechanisms that mediate the statin effect on insulin sensitivity. Statins may affect substrate delivery to insulin-sensitive tissues or modulate insulin-activated signaling cascades that mediate glucose uptake. Insulin increases skeletal muscle perfusion and substrate delivery by enhancing eNOS activity. As described previously, statins also increase eNOS expression, which may result in increased capillary recruitment and glucose disposal (60). Insulin activates a series of kinase cascades that involve PI3K and Akt, resulting in the translocation of glucose transporters to cell membrane and enhanced glucose uptake (60). This cascade is inhibited by circulating cytokines (TNF-{alpha} and IL-6) (60). Statins, like insulin, activate PI3K and Akt, which may play a role in glucose uptake. Statins, in addition to decreasing cytokine levels, also inhibit the cellular cascades such as Rho-kinase that inactivate the insulin receptor and signaling (20). NO is a potential intermediary, because it has been shown to stimulate skeletal muscle glucose uptake (61). Further studies (in vivo and in vitro) are needed to better understand the favorable effect of statin on glucose metabolism and insulin sensitivity.
continued...
|